 I am a computer scientist turned applied mathematician developing statistical methods to infer non-local properties of networks – like measures of long-range connectivity and causal effects – under realistic constraints of partial observations in the natural and social sciences. I am a Schmidt Science Fellow in the Institute for Data, Systems, and Society at Massachusetts Institute of Technology, working on causal inference in networked settings with Prof. Dean Eckles at MIT Sloan School of Management and Prof. Elchanan Mossel at MIT Mathematics.
I am a computer scientist turned applied mathematician developing statistical methods to infer non-local properties of networks – like measures of long-range connectivity and causal effects – under realistic constraints of partial observations in the natural and social sciences. I am a Schmidt Science Fellow in the Institute for Data, Systems, and Society at Massachusetts Institute of Technology, working on causal inference in networked settings with Prof. Dean Eckles at MIT Sloan School of Management and Prof. Elchanan Mossel at MIT Mathematics.
I was previously an EPSRC Doctoral Prize Fellow in the Department of Mathematics at Imperial College London, where I also completed my PhD supervised by Prof. Nick S. Jones in the Systems and Signals group at Imperial and Prof. Sumeet Agarwal at IIT Delhi. I studied how people form social connections, and how social networks relate to their health, which involves research at the crossroads of network science, sociology and public health. In particular, I developed methods to infer the structure and statistics of large-scale social networks. My talks at Networks 2021 and Sunbelt 2020, and poster presented at IC2S2, capture some of my research on these topics. I also collaborate with the Vaccine Confidence Project at LSHTM and Social-Decision Making Lab at Cambridge to study misinformation and its impact on the COVID-19 pandemic.
Prior to my PhD I was a post-baccalaureate fellow supervised by Prof. James J. Collins at the Wyss Institute for Biologically Inspired Engineering at Harvard University. I worked at the intersection of machine learning and biology, developing technologies for drug discovery, molecular diagnostics, and biological circuit design. I was fortunate enough to work alongside some great experimental biologists, which led to the development of computational tools such as NeMoCAD for drug discovery and MALDI spectral analysis for universal infection diagnosis. This talk I gave at the DAIR seminar at IIT Delhi is a summary of my pursuit to bring mathematical order to biology.
I did my B.Tech in Computer Science and Engineering at IIT Delhi, where my research was primarily guided by Prof. Sumeet Agarwal. My bachelors’ thesis focused on building causal models of gene regulatory networks.
My broader research interest is to apply mathematics and machine learning to better understand how complex biological, cognitive and eventually social systems work–and perhaps more importantly, when they don’t work. And in turn, use the knowledge gained to refine notions of computation itself. I’ve written more deeply about my thoughts on that here.
| Resume | GitHub | Email: firstinitiallastname@mit.edu |
Research Philosophy
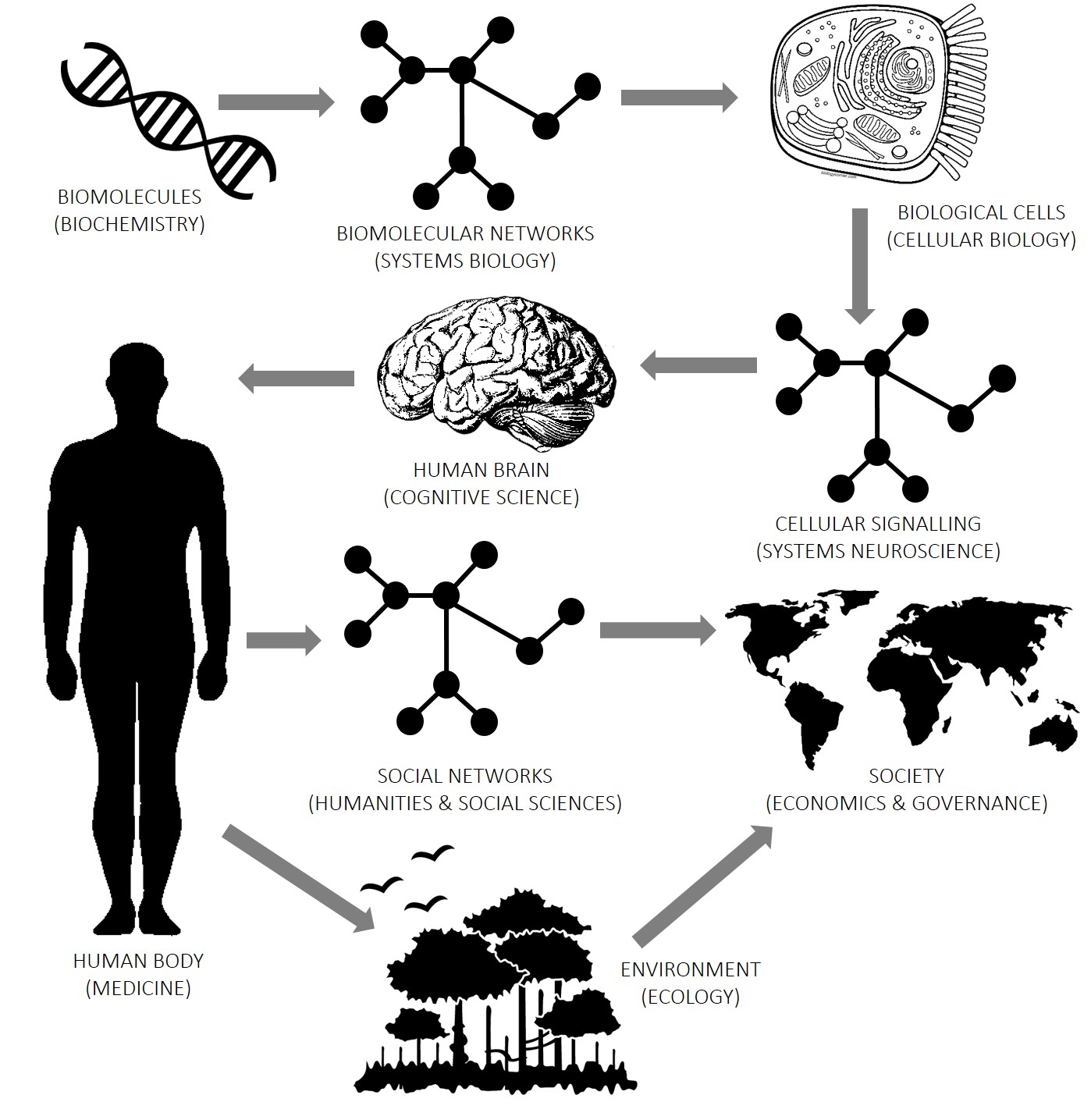 Computation is a vital tool for understanding things at all levels of organization of the world. Right from the fundamental particles of the universe, to the biomolecules that make life possible, to the wonders of biological intelligence, up to the intricate social interactions that create human civilization. There is a lot of shared structure of problems at various levels of the hierarchy, for instance the prevalance of graph structures at the biological, cognitive and societal levels, which is the recurring theme of my research. This pattern, when abstracted out, is precisely what permits the omnipotent use of computation.
Computation is a vital tool for understanding things at all levels of organization of the world. Right from the fundamental particles of the universe, to the biomolecules that make life possible, to the wonders of biological intelligence, up to the intricate social interactions that create human civilization. There is a lot of shared structure of problems at various levels of the hierarchy, for instance the prevalance of graph structures at the biological, cognitive and societal levels, which is the recurring theme of my research. This pattern, when abstracted out, is precisely what permits the omnipotent use of computation.
Below, I have attempted to lay out my research along this ladder of abstraction.
| Computation Core | For Biology | For Cognition | For Society |
Computation Core
Partially observed networks
Geodesic statistics for random network families
 A key task in the study of networked systems is to derive local and global properties that impact connectivity, synchronizability, and robustness. Computing shortest paths or geodesics in the network yields measures of node centrality and network connectivity that can contribute to explain such phenomena. We derive an analytic distribution of shortest path lengths, on the giant component in the supercritical regime or on small components in the subcritical regime, of any sparse (possibly directed) graph with conditionally independent edges, in the infinite-size limit. We provide specific results for widely used network families like stochastic block models, dot-product graphs, random geometric graphs, and graphons. The survival function of the shortest path length distribution possesses a simple closed-form lower bound which is asymptotically tight for finite lengths, has a natural interpretation of traversing independent geodesics in the network, and delivers novel insight in the above network families. Notably, the shortest path length distribution allows us to derive, for the network families above, important graph properties like the bond percolation threshold, size of the giant component, average shortest path length, and closeness and betweenness centralities. We also provide a corroborative analysis of a set of 20 empirical networks. This unifying framework demonstrates how geodesic statistics for a rich family of random graphs can be computed cheaply without having access to true or simulated networks, especially when they are sparse but prohibitively large
A key task in the study of networked systems is to derive local and global properties that impact connectivity, synchronizability, and robustness. Computing shortest paths or geodesics in the network yields measures of node centrality and network connectivity that can contribute to explain such phenomena. We derive an analytic distribution of shortest path lengths, on the giant component in the supercritical regime or on small components in the subcritical regime, of any sparse (possibly directed) graph with conditionally independent edges, in the infinite-size limit. We provide specific results for widely used network families like stochastic block models, dot-product graphs, random geometric graphs, and graphons. The survival function of the shortest path length distribution possesses a simple closed-form lower bound which is asymptotically tight for finite lengths, has a natural interpretation of traversing independent geodesics in the network, and delivers novel insight in the above network families. Notably, the shortest path length distribution allows us to derive, for the network families above, important graph properties like the bond percolation threshold, size of the giant component, average shortest path length, and closeness and betweenness centralities. We also provide a corroborative analysis of a set of 20 empirical networks. This unifying framework demonstrates how geodesic statistics for a rich family of random graphs can be computed cheaply without having access to true or simulated networks, especially when they are sparse but prohibitively large
| arXiv preprint | Networks 2021 talk | ↑ |
X-t-SNE
Data visualization of multiple high-D spaces with associated graph structures
 t-Stochastic Neighbor Embedding is a method of visualizing very high-dimensional data in 2 or 3 dimensions, that is now ubiquitiously used in data analysis across disciplines. The standard implementation allows visualization of a single feature space. We extend t-SNE to multiple feature spaces that could have an associated graph structure. That is, every data-point exists in multiple spaces, and all the data points are related in one or more graph structures. This extension is very useful since the generic graph structure can encode interesting domain knowledge that guides data visualization in the low-D space.
t-Stochastic Neighbor Embedding is a method of visualizing very high-dimensional data in 2 or 3 dimensions, that is now ubiquitiously used in data analysis across disciplines. The standard implementation allows visualization of a single feature space. We extend t-SNE to multiple feature spaces that could have an associated graph structure. That is, every data-point exists in multiple spaces, and all the data points are related in one or more graph structures. This extension is very useful since the generic graph structure can encode interesting domain knowledge that guides data visualization in the low-D space.
| slides | videos | ↑ |
iWMMM
Multimodal clustering of data by stacking DP and multioutput-GP priors
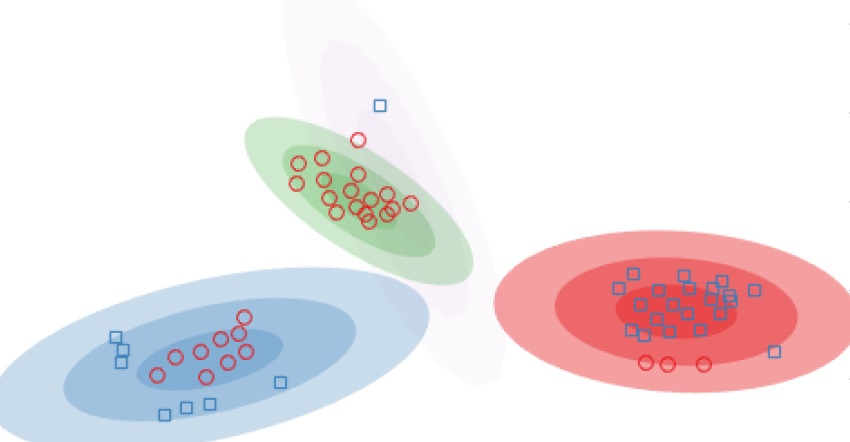 Gaussian mixture models are popular for clustering problems, where every cluster is assumed to be generated from a Gaussian distribution in the feature space. Placing a Dirichlet process prior on this space allows us to also automatically discover the number of clusters in the feature space, known as infinite Gaussian mixture models. However, the assumption of a Gaussian shaped cluster could be flawed. Instead, one can assume the feature space to be the output of a mapping from a latent space, where the Gaussian assumption might hold. Assuming a Gaussian process mapping between the latent and output spaces allows us to cluster arbitrarily warped clusters, called the infinite warped mixture model. We extend this approach to when we have multiple output feature spaces, assuming each to come from an independent Gaussian process from the same latent space.
Gaussian mixture models are popular for clustering problems, where every cluster is assumed to be generated from a Gaussian distribution in the feature space. Placing a Dirichlet process prior on this space allows us to also automatically discover the number of clusters in the feature space, known as infinite Gaussian mixture models. However, the assumption of a Gaussian shaped cluster could be flawed. Instead, one can assume the feature space to be the output of a mapping from a latent space, where the Gaussian assumption might hold. Assuming a Gaussian process mapping between the latent and output spaces allows us to cluster arbitrarily warped clusters, called the infinite warped mixture model. We extend this approach to when we have multiple output feature spaces, assuming each to come from an independent Gaussian process from the same latent space.
| slides | ↑ |
Hierarchical Ensemble Classifier
An ensemble technique for multi-label classification
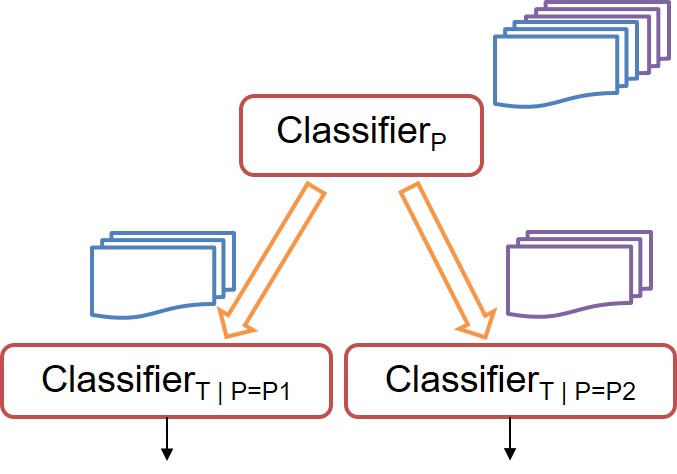 Many multi-classification problems have multiple output labels, wherein the same feature space has more than one output labels. The classification problem might be “easier” for some labels over others. With that intuition in mind, we develop a hierarchical metaclassifier which conditions single-label classifiers at lower levels on the class predicted for labels at the upper levels. That is, more difficult classification problems are conditioned over the easier ones. This flexible model can use any off-the-shelf multi-class single-label classifier at each of the levels. We test our algorithm on a two-label multi(2x4)-class problem of predicting the tolerance and pathogen of infection of a cohort of patients.
Many multi-classification problems have multiple output labels, wherein the same feature space has more than one output labels. The classification problem might be “easier” for some labels over others. With that intuition in mind, we develop a hierarchical metaclassifier which conditions single-label classifiers at lower levels on the class predicted for labels at the upper levels. That is, more difficult classification problems are conditioned over the easier ones. This flexible model can use any off-the-shelf multi-class single-label classifier at each of the levels. We test our algorithm on a two-label multi(2x4)-class problem of predicting the tolerance and pathogen of infection of a cohort of patients.
| slides | code | ↑ |
Computation for Biology
Biomolecules
NeMoCAD
Network Model for Causally Aware Discovery
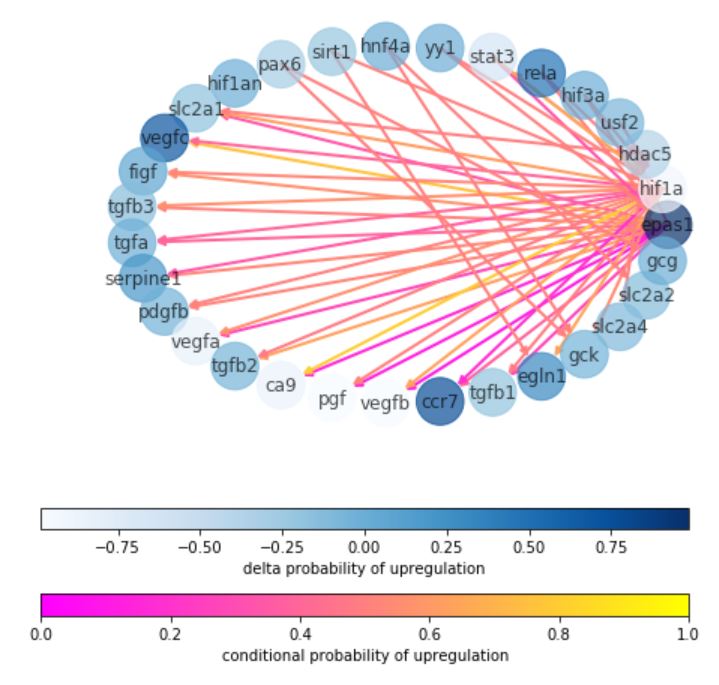 The problem of drug discovery can be seen as a problem of inducing a given model system, such as frog embryos, into a targetted transcriptomic state–a snapshot of which genes or proteins are “on” or “off” in the desired state. However, many genes interact with one another through regulatory mechanisms. Moreover, many drugs can have unintended side-effects since they impact more than one gene of interest. Therefore, we learn a network-aware Bayesian model from gene-gene and drug-gene datasets, which can be “queried” for appropriate gene therapies. We further extend this pipeline to affect not just desired genotypes, but phenotypes directly, which can facilitate closed-loop high-throughput drug screenings without performing transcriptomics analyses.
The problem of drug discovery can be seen as a problem of inducing a given model system, such as frog embryos, into a targetted transcriptomic state–a snapshot of which genes or proteins are “on” or “off” in the desired state. However, many genes interact with one another through regulatory mechanisms. Moreover, many drugs can have unintended side-effects since they impact more than one gene of interest. Therefore, we learn a network-aware Bayesian model from gene-gene and drug-gene datasets, which can be “queried” for appropriate gene therapies. We further extend this pipeline to affect not just desired genotypes, but phenotypes directly, which can facilitate closed-loop high-throughput drug screenings without performing transcriptomics analyses.
| slides | docs | notebook | code | ↑ |
Project THoR
Developing a probabilistic model of tolerance to pathogens in multiple host species
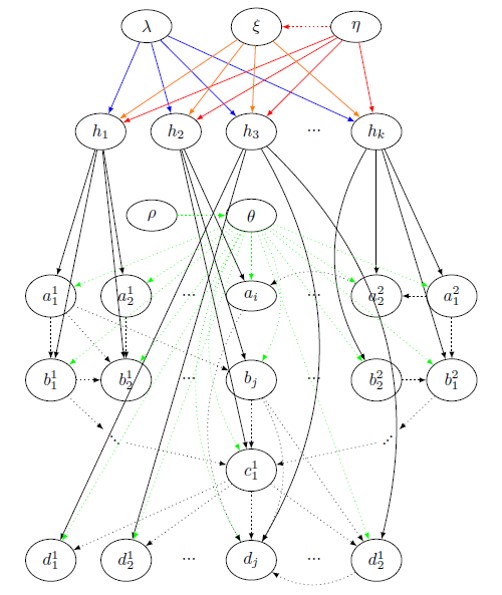 Given that most bacteria are becoming resistant to antibiotics, there is an urgent need to flip our practice of diagnosing and treating pathogen infections. Instead of exterminating the pathogen, we must start looking at ways to make a host “tolerant” to the infection, that is, stay asymptomatic despite infection, and develop Technologies for Host Resilience (THoR). For that, we look at the problem of predicting tolerance, and unearthing the underlying biological mechanisms of tolerance. We define a highly generalized Bayesian probabilistic framework called CROM3TOP (Cross-species Multimodal Modular Model of Tolerance to Pathogens) which can feed on (possibly temporal) multi-omics data across various host and pathogen species, to develop a rich ontology which not only differentiates between states of tolerance and sensitivity, but also provides a valuable interface for biologists to ask arbitrary queries of interest. This would help discover novel host-pathogen mechanisms, and hasten the design of gene therapies and other medical interventions.
Given that most bacteria are becoming resistant to antibiotics, there is an urgent need to flip our practice of diagnosing and treating pathogen infections. Instead of exterminating the pathogen, we must start looking at ways to make a host “tolerant” to the infection, that is, stay asymptomatic despite infection, and develop Technologies for Host Resilience (THoR). For that, we look at the problem of predicting tolerance, and unearthing the underlying biological mechanisms of tolerance. We define a highly generalized Bayesian probabilistic framework called CROM3TOP (Cross-species Multimodal Modular Model of Tolerance to Pathogens) which can feed on (possibly temporal) multi-omics data across various host and pathogen species, to develop a rich ontology which not only differentiates between states of tolerance and sensitivity, but also provides a valuable interface for biologists to ask arbitrary queries of interest. This would help discover novel host-pathogen mechanisms, and hasten the design of gene therapies and other medical interventions.
| code | ↑ |
Project SD2
Automating the experiment –> discover & design circuits –> test pipeline of synthetic biology
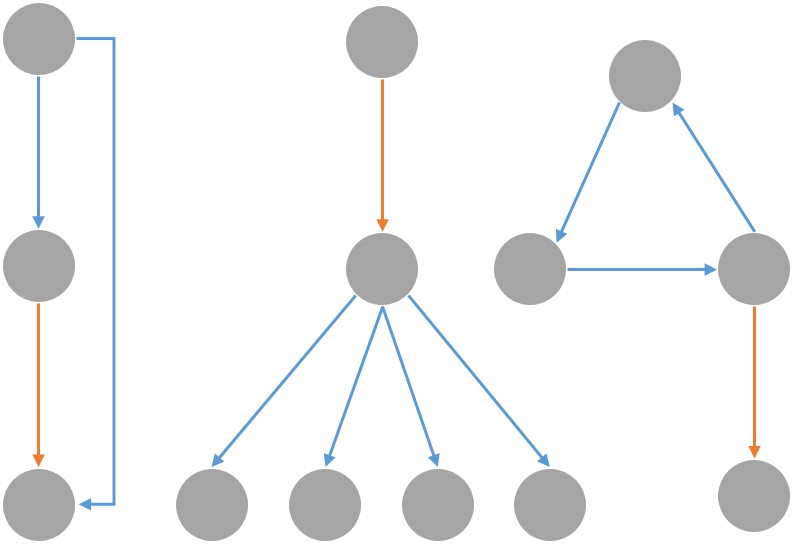 With the recent advent of synthetic biology, we are now engineering biological circuits to perform functions of our choosing, may it be doing arithmetic operations or diagnosing Zika. Since the space of functional circuits is huge, it has become increasingly important to automate the process of Synthetic Discovery and Design (SD2) of biological circuits. By making use of elementary logical motifs as the basic constituent of any complex function, we propose a novel algorithmic pipeline that goes from large-scale experimental data on biomolecular abundance in a system, to network structures (via network inference methods), to functional logical motifs (via graph embeddings such as node2vec and holographic embeddings), and eventually to operable and viable biological ciruits (via Bayesian model selection over biokentic ODE models). This would allow us to generate novel and testable scientific hypotheses for biologists to test in the lab, and will thus create a new cycle of experimental data that can be iteratively used to refine our models.
With the recent advent of synthetic biology, we are now engineering biological circuits to perform functions of our choosing, may it be doing arithmetic operations or diagnosing Zika. Since the space of functional circuits is huge, it has become increasingly important to automate the process of Synthetic Discovery and Design (SD2) of biological circuits. By making use of elementary logical motifs as the basic constituent of any complex function, we propose a novel algorithmic pipeline that goes from large-scale experimental data on biomolecular abundance in a system, to network structures (via network inference methods), to functional logical motifs (via graph embeddings such as node2vec and holographic embeddings), and eventually to operable and viable biological ciruits (via Bayesian model selection over biokentic ODE models). This would allow us to generate novel and testable scientific hypotheses for biologists to test in the lab, and will thus create a new cycle of experimental data that can be iteratively used to refine our models.
| slides | code | ↑ |
protein2vec
Embedding peptide sequences into a low-D vector space
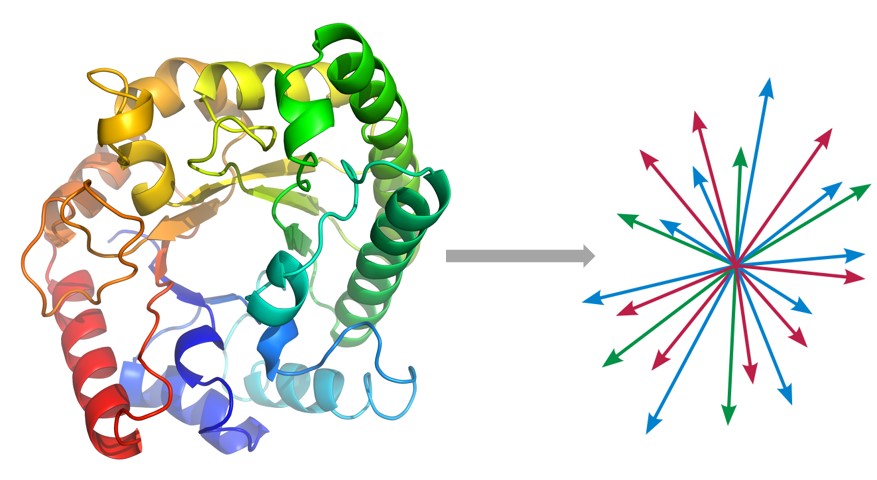 We are still far from solving a lot of problems in proteomics, may it be predicting protein folding structure, or their binding characterization, which are key to fully understanding how proteins participate in key biochemical reactions that define life as we know it. In collaboration with the Church group, we define an unsupervised problem of embedding protein sequences into an appropriate vector space, so that we can apply statistical machine learning for supervised problem of interest. We are building a seq2seq model of peptide sequences, based on the RNN encoder-decoder and Google’s attention-based Transformer model. Besides encoding the primary sequence, we would concomitantly encode relevant physicochemical properties of amino acids that can produce more biologically meaningful embeddings.
We are still far from solving a lot of problems in proteomics, may it be predicting protein folding structure, or their binding characterization, which are key to fully understanding how proteins participate in key biochemical reactions that define life as we know it. In collaboration with the Church group, we define an unsupervised problem of embedding protein sequences into an appropriate vector space, so that we can apply statistical machine learning for supervised problem of interest. We are building a seq2seq model of peptide sequences, based on the RNN encoder-decoder and Google’s attention-based Transformer model. Besides encoding the primary sequence, we would concomitantly encode relevant physicochemical properties of amino acids that can produce more biologically meaningful embeddings.
| code | ↑ |
Gaussian Process Models for Time-Series Omics Analysis
Probabilistic modeling of gene expression evolution with time
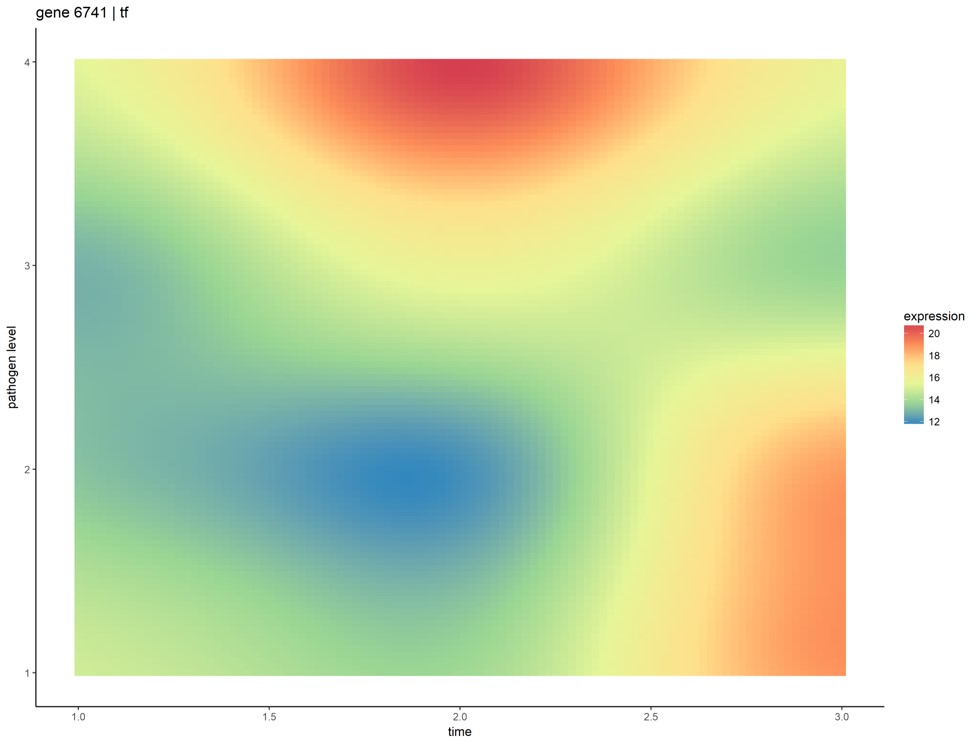 Most biological systems have associated temporal dynamics, which are key to their complete mechanistic understanding. Regulation and expression of genes, their translation into proteins, and their consequent effects on biochemical reactions, are events unfolding in accordance with their respective kinetics. Current omics analyses for comparing samples across different phenotypes, like differential expression, focus on single snapshots of a system’s state (either at a particular point in or averaged over time). We applied two approaches to principally model time-series omics data using Gaussian process regression, and introduced methods for comparative analysis of multiple phenotypes with only few samples per phenotype. We used transcriptional data of frog embryos infected with four different initial doses of Pseudomonas aeruginosa, collected over the first 3 days of development. Key gene groups, that relate to pathways and processes involved in host-pathogen interactions, were also identified.
Most biological systems have associated temporal dynamics, which are key to their complete mechanistic understanding. Regulation and expression of genes, their translation into proteins, and their consequent effects on biochemical reactions, are events unfolding in accordance with their respective kinetics. Current omics analyses for comparing samples across different phenotypes, like differential expression, focus on single snapshots of a system’s state (either at a particular point in or averaged over time). We applied two approaches to principally model time-series omics data using Gaussian process regression, and introduced methods for comparative analysis of multiple phenotypes with only few samples per phenotype. We used transcriptional data of frog embryos infected with four different initial doses of Pseudomonas aeruginosa, collected over the first 3 days of development. Key gene groups, that relate to pathways and processes involved in host-pathogen interactions, were also identified.
| slides | ↑ |
Project ConDDR
Drug repurposing using an information-retrieval strategy
 Thousands of medically approved drugs are currently used to treat various diseases, but they can have complex interactions with more than one receptor proteins. That is, the same drug can interact with multiple receptors and thus be repurposed for treating a different disease. Moreover, their combinations can be given as so-called “drug cocktails” to improve a treatment’s efficacy, as is now the norm in cancer therapies. With this motivation in mind we propose Context Dependent Drug Repurposing. We extracted an ontology of drug-gene interactions. We employ techniques of information retrieval on this interaction matrix, by treating it as a document-word matrix and genes-of-interest as a query vector. We successfully “rediscover” conventional drugs for diseases such as TB, and discover novel ones for other bacterial infections.
Thousands of medically approved drugs are currently used to treat various diseases, but they can have complex interactions with more than one receptor proteins. That is, the same drug can interact with multiple receptors and thus be repurposed for treating a different disease. Moreover, their combinations can be given as so-called “drug cocktails” to improve a treatment’s efficacy, as is now the norm in cancer therapies. With this motivation in mind we propose Context Dependent Drug Repurposing. We extracted an ontology of drug-gene interactions. We employ techniques of information retrieval on this interaction matrix, by treating it as a document-word matrix and genes-of-interest as a query vector. We successfully “rediscover” conventional drugs for diseases such as TB, and discover novel ones for other bacterial infections.
Causal Computational Models for GRNs
Developiong a causal computational model for gene regulatory networks
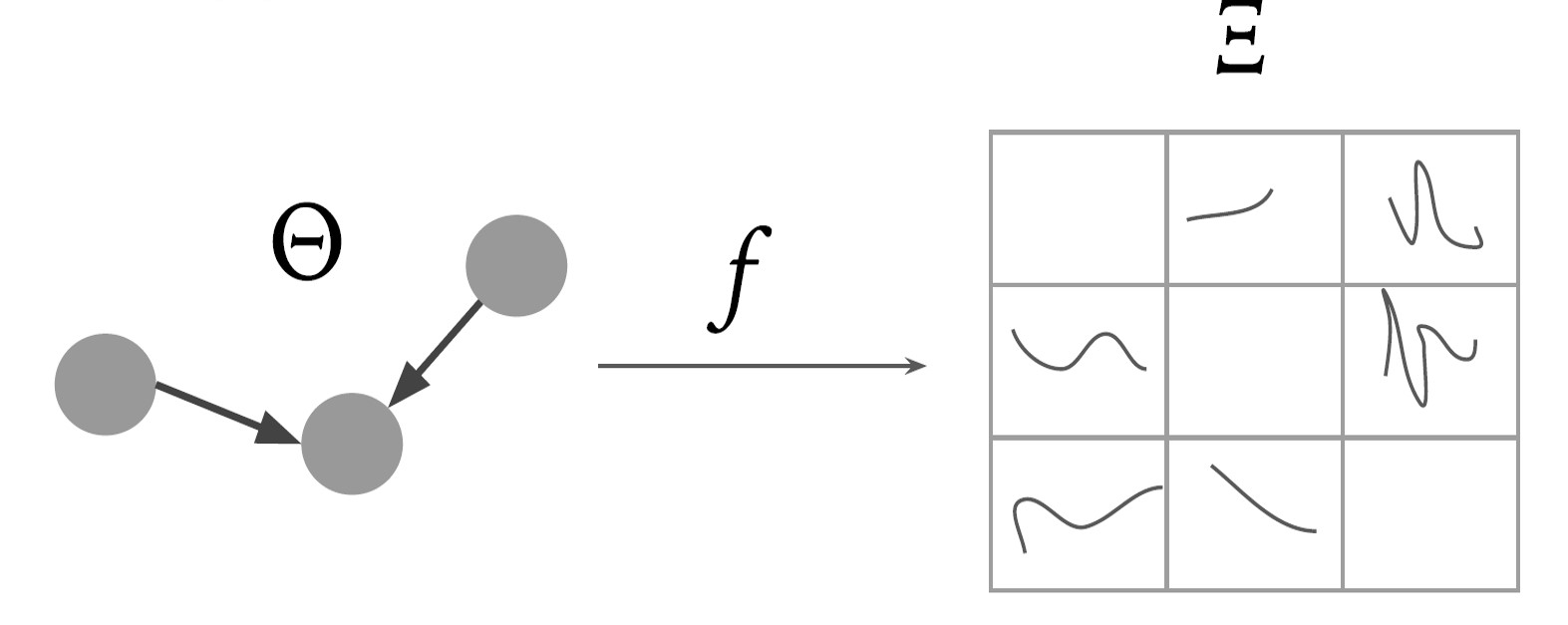 Gene Regulatory Networks (GRNs) hold the key to understanding and solving many problems in biological sciences, with critical applications in medicine and therapeutics. However, discovering GRNs in the laboratory is a cumbersome and tricky affair, since the number of genes and interactions, say in a mammalian cell, are very large. We aim to discover these GRNs computationally, by using gene expression levels as a time-series dataset. We research and employ techniques from probability and information theory, theory of dynamical systems, to establish pairwise causal relations between genes on synthetic datasets. Furthermore, we suggest methods for global estimation of gene networks by using intrinsic graph estimation and a random-walk based weight propagation algorithm.
Gene Regulatory Networks (GRNs) hold the key to understanding and solving many problems in biological sciences, with critical applications in medicine and therapeutics. However, discovering GRNs in the laboratory is a cumbersome and tricky affair, since the number of genes and interactions, say in a mammalian cell, are very large. We aim to discover these GRNs computationally, by using gene expression levels as a time-series dataset. We research and employ techniques from probability and information theory, theory of dynamical systems, to establish pairwise causal relations between genes on synthetic datasets. Furthermore, we suggest methods for global estimation of gene networks by using intrinsic graph estimation and a random-walk based weight propagation algorithm.
| slides 1 | slides 2 | slides 3 | slides 4 | code | ↑ |
Evolvable Networks
Evolving synthetic biological networks using evolutionary principles
 Biological networks, may they be gene regulatory networks or protein-protein interaction networks, tend to possess certain attributes. They are highly modular, seem to be very robust, and also appear to be scale-free. Thus, one might be tempted to believe that to create instances of biological networks, one must explicate such conditions on a graph. However, we hypothesize that these are merely emergent properties which are not evidently very principled. Rather, a certain sense of “evolvability” (E), combined with “robustness” (R), and a “cost of complexity” (C) levied on the network, seem to be principled evolutionary factors due to which biological networks might organize themselves. Armed with this intuition, we evolve biological networks by incorporating ERC into a genetic algorithm’s objective function. The networks so obtained appear to observe these emergent properties.
Biological networks, may they be gene regulatory networks or protein-protein interaction networks, tend to possess certain attributes. They are highly modular, seem to be very robust, and also appear to be scale-free. Thus, one might be tempted to believe that to create instances of biological networks, one must explicate such conditions on a graph. However, we hypothesize that these are merely emergent properties which are not evidently very principled. Rather, a certain sense of “evolvability” (E), combined with “robustness” (R), and a “cost of complexity” (C) levied on the network, seem to be principled evolutionary factors due to which biological networks might organize themselves. Armed with this intuition, we evolve biological networks by incorporating ERC into a genetic algorithm’s objective function. The networks so obtained appear to observe these emergent properties.
| slides | ↑ |
Organisms
Project MALDI for Diagnosis
A probabilistic model for detecting pathogens in a given sample
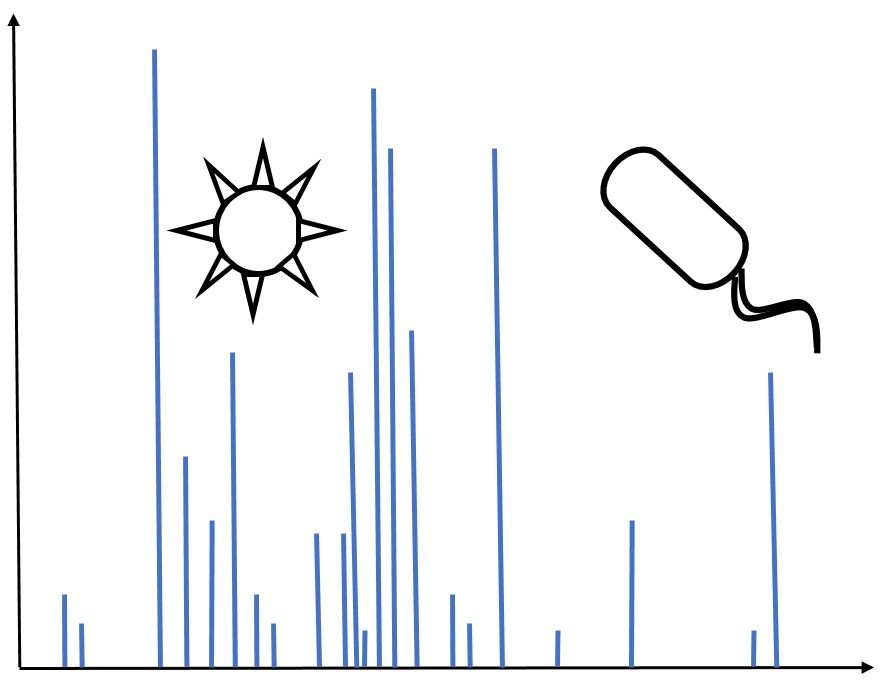 The purpose of this project was to identify Mycobacterium tuberculosis within biological fluids, particularly human urine, for use as a diagnostic. We have extracted pathogen from solutions using FcMBL magnetic nanobead technology. The captured material is then analyzed using matrix-assisted laser deionization time-of-flight/time-of-flight (MALDI-TOF/TOF) tandem mass spectrometry. This produces a series of peaks specifying the mass-to-charge ratios of the sample of interest. Peak libraries exclusive to several strains of M. tuberculosis as well as other microbes including Staphylococcus aureus and Candida albicans have been assembled. We created a probabilistic model from these libraries in order to identify a pathogen from an unknown sample.
The purpose of this project was to identify Mycobacterium tuberculosis within biological fluids, particularly human urine, for use as a diagnostic. We have extracted pathogen from solutions using FcMBL magnetic nanobead technology. The captured material is then analyzed using matrix-assisted laser deionization time-of-flight/time-of-flight (MALDI-TOF/TOF) tandem mass spectrometry. This produces a series of peaks specifying the mass-to-charge ratios of the sample of interest. Peak libraries exclusive to several strains of M. tuberculosis as well as other microbes including Staphylococcus aureus and Candida albicans have been assembled. We created a probabilistic model from these libraries in order to identify a pathogen from an unknown sample.
| webapp | code | manuscript in prep | ↑ |
Game of Apoptosis
A Boolean network analysis of the tightly-regulated apoptosis pathway
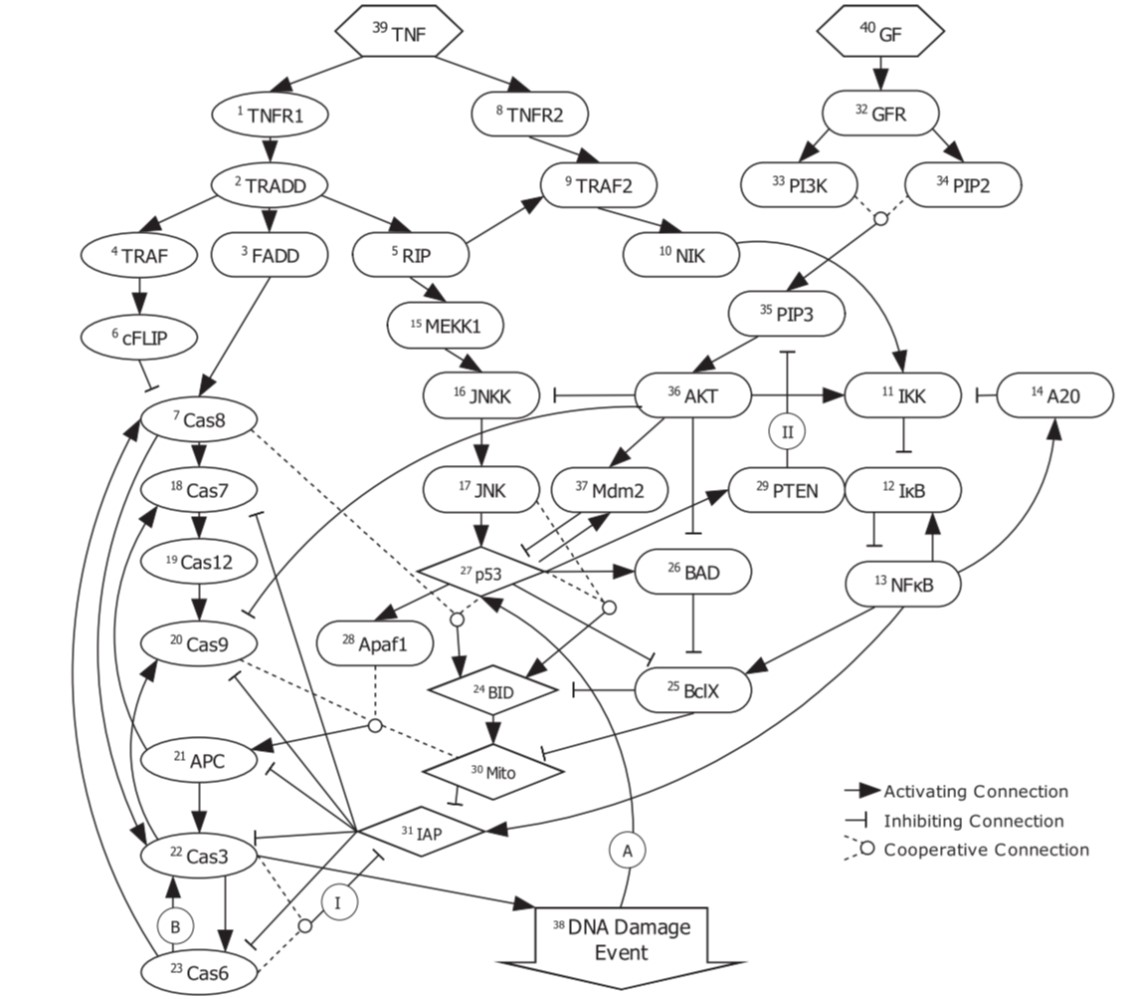 Apoptosis refers to one of the many biochemical process that take place inside a cell. Known as “programmed cell death”, it leads to fatal changes in the cell such as decay and DNA fragmentation. It is a highly controlled and regulated process, whose understanding can throw light on critical diseases such as cancer (under-regulated apoptosis leads to cell proliferation) and atrophy (over-regulated apoptosis leads to excessive cell death). There are multiple ways to understand regulatory networks, such as using ordinary differential equations, or using graphical methods like Bayesian and Boolean networks. The latter capture basic activating/inhibitory relationship between genes being in an on-off state (0/1, thus Boolean). We analyze one such Boolean model given by Mai et al. (2009), devise metrics of studying the irreversibility of apoptosis, and improve the analysis of which initial cell states are more likely to lead to apoptosis.
Apoptosis refers to one of the many biochemical process that take place inside a cell. Known as “programmed cell death”, it leads to fatal changes in the cell such as decay and DNA fragmentation. It is a highly controlled and regulated process, whose understanding can throw light on critical diseases such as cancer (under-regulated apoptosis leads to cell proliferation) and atrophy (over-regulated apoptosis leads to excessive cell death). There are multiple ways to understand regulatory networks, such as using ordinary differential equations, or using graphical methods like Bayesian and Boolean networks. The latter capture basic activating/inhibitory relationship between genes being in an on-off state (0/1, thus Boolean). We analyze one such Boolean model given by Mai et al. (2009), devise metrics of studying the irreversibility of apoptosis, and improve the analysis of which initial cell states are more likely to lead to apoptosis.
| slides | code | ↑ |
Project Abbie
An ML model for predicting the severity of breathing under an asthma attack
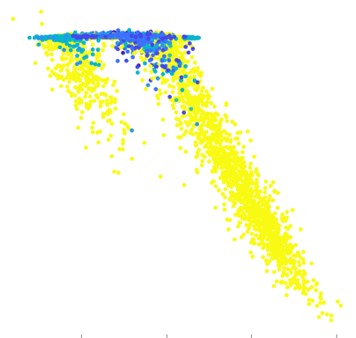 Asthma is a common inflammatory disease of lung airways which affects millions of people across the world. An asthmatic fit can be triggered by allergens and is accompanied by symptoms such as wheezing, shortness of breath and chest spasms. A timely prediction of the fit can allow an asthmatic to seek help and administer a response on time, which could be life saving. As part of Project Abbie, in collaboration with Boston Children’s Hospital, we collected breathing waveform data of children from the beginning to end of an asthmatic fit, and the corresponding severity score through this timecourse. We extracted a multifractal spectrum representation of the signal; hypothesized and validated that the onset of an attack is accompanied by a change from mono to multifracticality of the breathing waveform. We built a simple 2-class model to predict a high/low severity score of an asthmatic over a 10-second time window, which can allow real time monitoring of patients.
Asthma is a common inflammatory disease of lung airways which affects millions of people across the world. An asthmatic fit can be triggered by allergens and is accompanied by symptoms such as wheezing, shortness of breath and chest spasms. A timely prediction of the fit can allow an asthmatic to seek help and administer a response on time, which could be life saving. As part of Project Abbie, in collaboration with Boston Children’s Hospital, we collected breathing waveform data of children from the beginning to end of an asthmatic fit, and the corresponding severity score through this timecourse. We extracted a multifractal spectrum representation of the signal; hypothesized and validated that the onset of an attack is accompanied by a change from mono to multifracticality of the breathing waveform. We built a simple 2-class model to predict a high/low severity score of an asthmatic over a 10-second time window, which can allow real time monitoring of patients.
| slides | ↑ |
Computation for Cognition
Human Category Learning
Testing the brain lateralization of two models of human category learning
 Human cognitive systems rely heavily on how knowledge is represented within these systems. One kind of knowledge, namely the categorical, is ubiquitous in everyday human interaction with the world. Two competing theories of how humans learn and infer from category knowledge have been popular in cognitive psychology: the exemplar theory and the prototype theory. Also, like most cognitive functions, category representations have purported to be lateralized in the brain: with the left hemisphere operating under prototype conditions, and the right hemisphere operating under exemplar conditions. We explore the hypothesis as to whether category knowledge representation is indeed lateralized in the light of these theories, and if yes, then whether a transfer of control between the two gets activated at some epoch of learning. We conduct divided visual field tasks to collect data, and use Bayesian model selection to test our hypothesis.
Human cognitive systems rely heavily on how knowledge is represented within these systems. One kind of knowledge, namely the categorical, is ubiquitous in everyday human interaction with the world. Two competing theories of how humans learn and infer from category knowledge have been popular in cognitive psychology: the exemplar theory and the prototype theory. Also, like most cognitive functions, category representations have purported to be lateralized in the brain: with the left hemisphere operating under prototype conditions, and the right hemisphere operating under exemplar conditions. We explore the hypothesis as to whether category knowledge representation is indeed lateralized in the light of these theories, and if yes, then whether a transfer of control between the two gets activated at some epoch of learning. We conduct divided visual field tasks to collect data, and use Bayesian model selection to test our hypothesis.
| slides | webapp | code | ↑ |
Human Form Learning
A topological perspective on form learning in humans
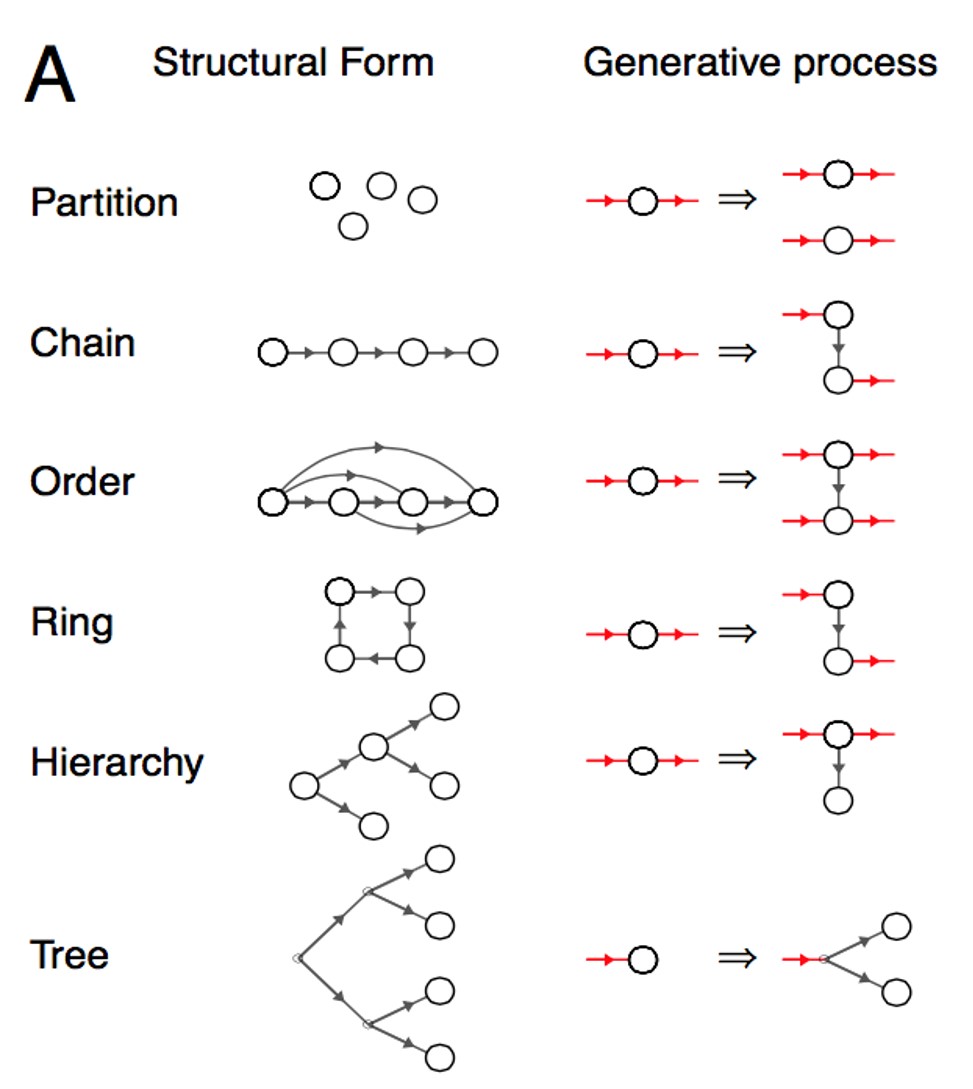 Given the resource constraints that human cognitive systems operate under, how do adults and children inductively learn from the percepts we encounter? Certainly, there are some heuristics involved in not just our inductive learning, but also in the way we do inference and reasoning. Can we explain away the use of such “shortcuts” by improving current computational cognitive models? This project was an attempt in answering such questions, by taking the work by Kemp et al. (2008) on form learning as a neat illustration of some key ideas. We extend their work by stressing on the need to include topology (manifold learning) and hierarchical Bayesian modelling, by providing experimental results to support our stance.
Given the resource constraints that human cognitive systems operate under, how do adults and children inductively learn from the percepts we encounter? Certainly, there are some heuristics involved in not just our inductive learning, but also in the way we do inference and reasoning. Can we explain away the use of such “shortcuts” by improving current computational cognitive models? This project was an attempt in answering such questions, by taking the work by Kemp et al. (2008) on form learning as a neat illustration of some key ideas. We extend their work by stressing on the need to include topology (manifold learning) and hierarchical Bayesian modelling, by providing experimental results to support our stance.
| slides | ↑ |
Human Motor Control
An ML model for predicting thought of motor activity using EEG signals
 Recent advances in modern brain-imaging techniques have hugely bolstered research in areas of brain sciences and cognitive studies. While they were earlier being used for merely diagnosing brain disorders, they are now being used to generate highly utilitarian bits of data. One such imaging technique is EEG, which essentially measures the averaged electrical potential across the human scalp. The motivation behind this project was to develop a computational basis for developing real-time brain controlled actions, such as thought-controlled prosthetic limbs or electronic devices. Can we recover signatures of motor activity, or the thought of motor activity, from the EEG signal? We conducted controlled experiments to collect EEG data. Using techniques from machine learning, we then classify them into three motion states of rest, arm jerk, and thought of arm jerk.
Recent advances in modern brain-imaging techniques have hugely bolstered research in areas of brain sciences and cognitive studies. While they were earlier being used for merely diagnosing brain disorders, they are now being used to generate highly utilitarian bits of data. One such imaging technique is EEG, which essentially measures the averaged electrical potential across the human scalp. The motivation behind this project was to develop a computational basis for developing real-time brain controlled actions, such as thought-controlled prosthetic limbs or electronic devices. Can we recover signatures of motor activity, or the thought of motor activity, from the EEG signal? We conducted controlled experiments to collect EEG data. Using techniques from machine learning, we then classify them into three motion states of rest, arm jerk, and thought of arm jerk.
| poster | slides | ↑ |
Complexity of Living and Biological Systems
On the limits to understanding complex systems, from a computational complexity POV
 The central objective of recent strides taken by biology and biochemistry has been to simplify the complexity of biological systems, and of life itself. The theories and models we propose, and the questions we try to answer, are subject to the current limits of our mathematics and computational abilities. There are, of course, various problems in computer science that have been deemed unsolvable (like the Halting Problem), or computationally intensive (like the Maximum Independent Set Problem which is NP hard), but can similar limits be imposed on problems in biology? Our intuition tells us yes, and recent advances in the theory of computation, and the principles of Computational Equivalence and Computational Irreducibility are in overwhelming support of this intuition. We follow these principles and view systems under the lens of computational pragmatism, to uncover the possible limits imposed on our understanding of life, the way we know it.
The central objective of recent strides taken by biology and biochemistry has been to simplify the complexity of biological systems, and of life itself. The theories and models we propose, and the questions we try to answer, are subject to the current limits of our mathematics and computational abilities. There are, of course, various problems in computer science that have been deemed unsolvable (like the Halting Problem), or computationally intensive (like the Maximum Independent Set Problem which is NP hard), but can similar limits be imposed on problems in biology? Our intuition tells us yes, and recent advances in the theory of computation, and the principles of Computational Equivalence and Computational Irreducibility are in overwhelming support of this intuition. We follow these principles and view systems under the lens of computational pragmatism, to uncover the possible limits imposed on our understanding of life, the way we know it.
| ↑ |
Computation for Society
Social Systems
Learning Social Connectivity Kernels
A cheap method to infer social connectivity models from egocentric data
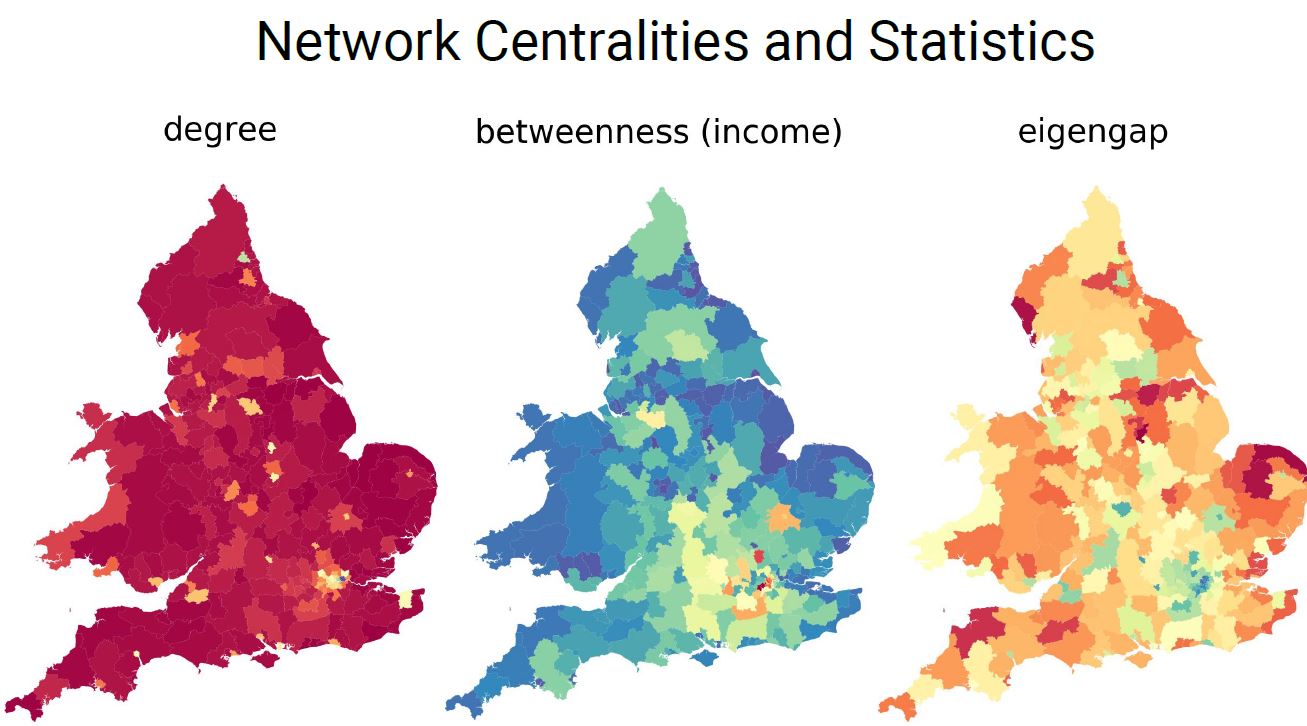 Social networks play a crucial role in determining social outcomes, particularly those related to people’s health and well-being. We present a novel method to learn probabilistic connectivity kernels from small-scale egocentric surveys, and consequently extract social access statistics from widely available socio-demographic surveys like the census. We demonstrate how different network centralities, that capture varied dimensions of social capital, correlate to people’s subjective and objective well-being. Being a generative model, representative networks can be sampled to pursue downstream social network analyses, both on network ensembles and at the model level.
Social networks play a crucial role in determining social outcomes, particularly those related to people’s health and well-being. We present a novel method to learn probabilistic connectivity kernels from small-scale egocentric surveys, and consequently extract social access statistics from widely available socio-demographic surveys like the census. We demonstrate how different network centralities, that capture varied dimensions of social capital, correlate to people’s subjective and objective well-being. Being a generative model, representative networks can be sampled to pursue downstream social network analyses, both on network ensembles and at the model level.
| code | notebook | manuscript in prep | ↑ |
Urban Mobility
How far would you go? Comparing urban access in 10 Global Cities
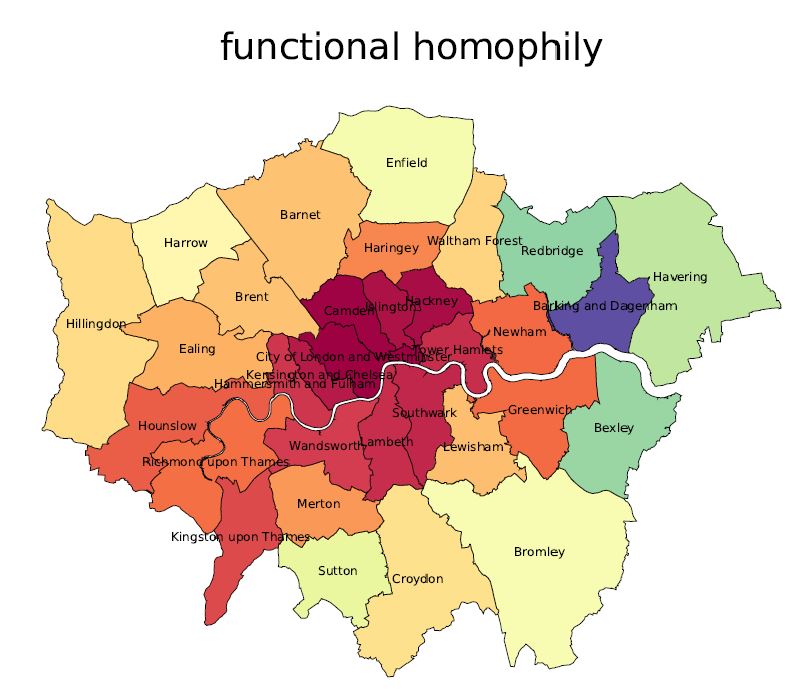 Cities permit people to access a diverse range of venues and attractions with relative ease. A range of factors can determine how successfully they do so. We provide a probabilistic framework that captures 3 inter-related yet independently varying views of urban mobility: spatial, functional and social. The spatial view captures number of public venues across physical space (say a city), and thus the opportunities of access. The functional view captures venues across functional categories (say transport, restaurants), and thus the type of access. The social view is the sum of all behavioural
considerations (people tend to go from offices to metro stations). We develop principled measures of urban and spatial access which are validated by using data on trip check-ins from the location-based social network Foursquare, on 10 cities spread across the globe. Analysis reveals that people travel further for venue diversity, and for venue types they have a high affinity for. Consequently, there is a trade-off between local venue diversity and global venue popularity.
Cities permit people to access a diverse range of venues and attractions with relative ease. A range of factors can determine how successfully they do so. We provide a probabilistic framework that captures 3 inter-related yet independently varying views of urban mobility: spatial, functional and social. The spatial view captures number of public venues across physical space (say a city), and thus the opportunities of access. The functional view captures venues across functional categories (say transport, restaurants), and thus the type of access. The social view is the sum of all behavioural
considerations (people tend to go from offices to metro stations). We develop principled measures of urban and spatial access which are validated by using data on trip check-ins from the location-based social network Foursquare, on 10 cities spread across the globe. Analysis reveals that people travel further for venue diversity, and for venue types they have a high affinity for. Consequently, there is a trade-off between local venue diversity and global venue popularity.
| paper | ↑ |
Rationality in Economics
Studying the unfounded assumptions of rationality in microeconomic theory
 The most foundational assumption in all of microeconomic theory is that of rationality: Homo economicus is an agent who is actuated only by self-interest. But there has been a lot of criticism for this key assumption, with counterexamples in the form of tragedy of the commons, and Sen’s Liberal Paradox. We explore the “rationality” of this human economic agent, motivated by Daniel Hausman’s anthology of essays “Philosophy of Economics” wherein he urges economists to not trust in the success of economic theories just because they seem to work, but to “look under the hood”. We look at arguments by Hirschman (against parsimony), Gneezy & Rustichini (against ceteris paribus) and Benabou & Tirole (for inclusion of social norms). Sen’s seminal paper, Rational Fools captures a similar sentiment into the idea of meta-preferences over usual preferences, ranked by laws and norms of the society. We propose an alternative look at economic theory, wherein theory is not founded on irrational assumptions of rationality. Rather, it is a constantly updated set of theories, driven by a previous iteration of policy making and public reaction.
The most foundational assumption in all of microeconomic theory is that of rationality: Homo economicus is an agent who is actuated only by self-interest. But there has been a lot of criticism for this key assumption, with counterexamples in the form of tragedy of the commons, and Sen’s Liberal Paradox. We explore the “rationality” of this human economic agent, motivated by Daniel Hausman’s anthology of essays “Philosophy of Economics” wherein he urges economists to not trust in the success of economic theories just because they seem to work, but to “look under the hood”. We look at arguments by Hirschman (against parsimony), Gneezy & Rustichini (against ceteris paribus) and Benabou & Tirole (for inclusion of social norms). Sen’s seminal paper, Rational Fools captures a similar sentiment into the idea of meta-preferences over usual preferences, ranked by laws and norms of the society. We propose an alternative look at economic theory, wherein theory is not founded on irrational assumptions of rationality. Rather, it is a constantly updated set of theories, driven by a previous iteration of policy making and public reaction.
| slides | readings | ↑ |
Wisdom of Crowds
A large-scale game to investigate the wisdom of crowds
 Wisdom of Crowds refers to the idea that the aggregated opinion of a crowd of non-experts could be as close or even better than the estimate of an expert. People from across disciplines, cognitive scientists, social scientists, computer scientists and mathematicians, are trying hard to test the validity of this conjecture. In the hopes of elucidating conditions under which it holds well, areas where this can be applied, and the relation of an individual’s cognitive abilities to the judgement of the crowd. As part of a large international team of students, we developed a large-scale online game to systematically investigate the wisdom of crowds.
Wisdom of Crowds refers to the idea that the aggregated opinion of a crowd of non-experts could be as close or even better than the estimate of an expert. People from across disciplines, cognitive scientists, social scientists, computer scientists and mathematicians, are trying hard to test the validity of this conjecture. In the hopes of elucidating conditions under which it holds well, areas where this can be applied, and the relation of an individual’s cognitive abilities to the judgement of the crowd. As part of a large international team of students, we developed a large-scale online game to systematically investigate the wisdom of crowds.
| paper | webapp | ↑ |
Pedagogy in the Contemporary World
Multimodal Table of Content for Videos
Automatic generation of a table of content for video lectures
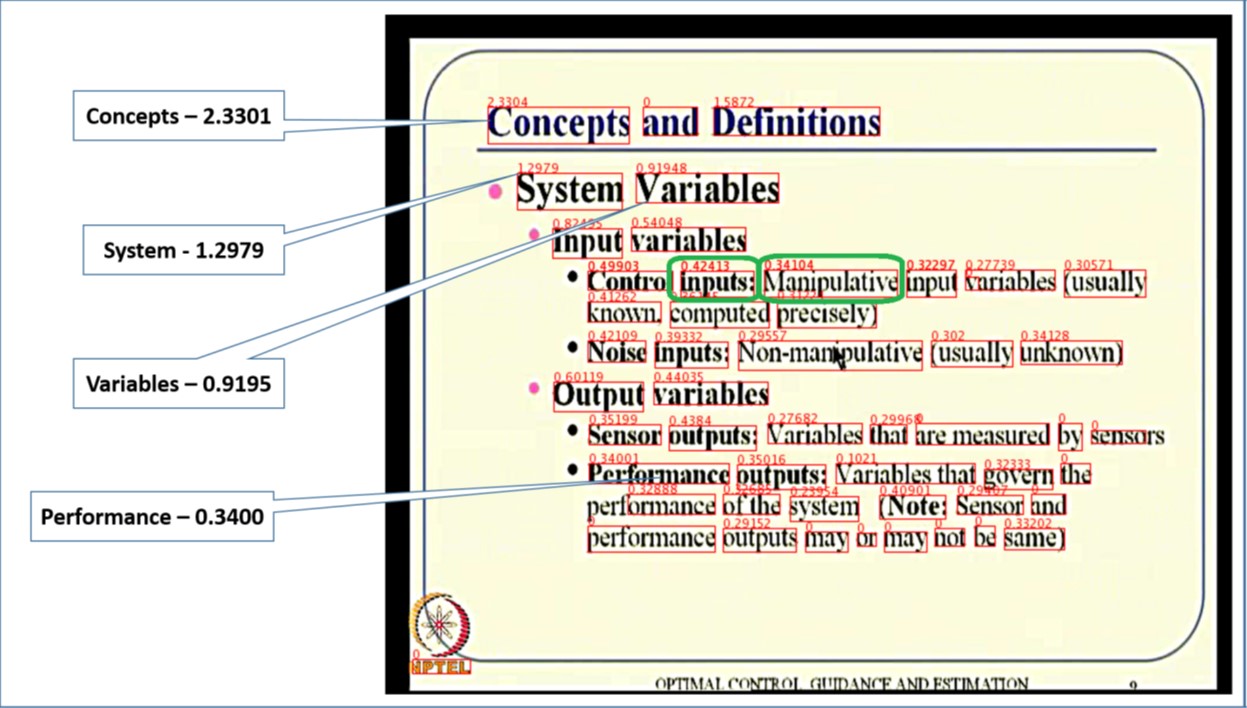 The amount of instructional videos available online is growing steadily. A major bottleneck in their widespread usage is the lack of tools for easy consumption of these videos. We developed MMToC (Multimodal Method for Table of Content), that automatically generates a table of content for a given instructional video and enables textbook-like efficient navigation through the video. MMToC quantifies word saliency for visual words extracted from the slides and for spoken words obtained from the lecture transcript. These saliency scores are combined using a segmentation algorithm to identify likely breakpoints in the video where the topic has changed.
The amount of instructional videos available online is growing steadily. A major bottleneck in their widespread usage is the lack of tools for easy consumption of these videos. We developed MMToC (Multimodal Method for Table of Content), that automatically generates a table of content for a given instructional video and enables textbook-like efficient navigation through the video. MMToC quantifies word saliency for visual words extracted from the slides and for spoken words obtained from the lecture transcript. These saliency scores are combined using a segmentation algorithm to identify likely breakpoints in the video where the topic has changed.
| paper | ↑ |
Video Lecture Sequencing
Automatic creation of a video-based curriculum for a given learning goal
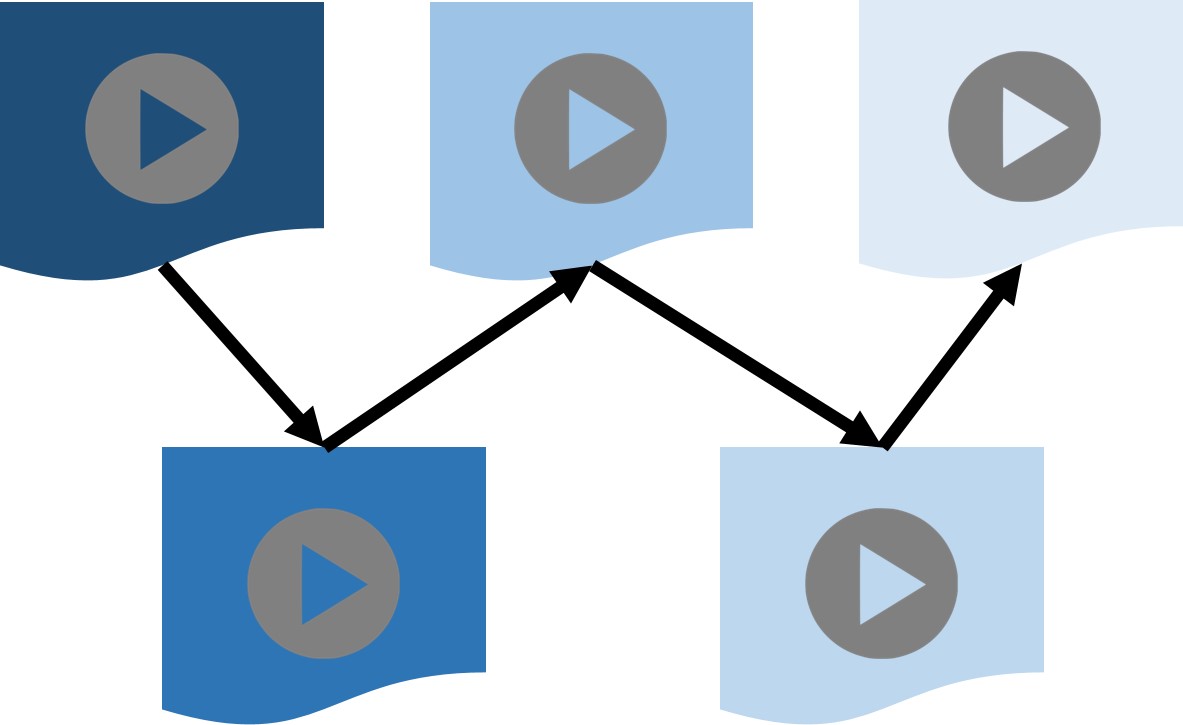 With a plethora of video lectures available for learning various topics, the cognitive burden on a new student learner can be very high in terms of (a) selecting the right video lectures to view for learning a topic of interest, and (b) charting out an appropriate video-based curriculum. We created an algorithm which extracts key concepts from video lectures, figures out an appropriate sequence of video lectures that respects the learning trajectory from pre-requisite concepts to learnt concepts, and charts out the shortest curriculum for a given learning goal while minimizing the cognitive burden on the learner.
With a plethora of video lectures available for learning various topics, the cognitive burden on a new student learner can be very high in terms of (a) selecting the right video lectures to view for learning a topic of interest, and (b) charting out an appropriate video-based curriculum. We created an algorithm which extracts key concepts from video lectures, figures out an appropriate sequence of video lectures that respects the learning trajectory from pre-requisite concepts to learnt concepts, and charts out the shortest curriculum for a given learning goal while minimizing the cognitive burden on the learner.
| code | ↑ |
Morality of Unsettling Interventions
A cognitive solution to the moral problem of unsettlement of core beliefs
 Unsettlement of our personal fundamental beliefs is a common occurrence in human interactions that allows people to update their beliefs about the world. This interventionary process can eventually define collective choice of a people. But it also places an interesting moral dilemma on the participants of the intervention: “is it right to unsettle someone else’s core beliefs?” and “should I let myself get unsettled?” We present an abstraction of the cognitive machinery of the agents involved in this moral problem, the way they represent knowledge and beliefs, and offer a way to resolve this dilemma for both the unsettler and the one unsettled.
Unsettlement of our personal fundamental beliefs is a common occurrence in human interactions that allows people to update their beliefs about the world. This interventionary process can eventually define collective choice of a people. But it also places an interesting moral dilemma on the participants of the intervention: “is it right to unsettle someone else’s core beliefs?” and “should I let myself get unsettled?” We present an abstraction of the cognitive machinery of the agents involved in this moral problem, the way they represent knowledge and beliefs, and offer a way to resolve this dilemma for both the unsettler and the one unsettled.
| slides | ↑ |
Environment
Plankton and Ocean Health
An ML model for classification of 120 plankton to quantify ocean health
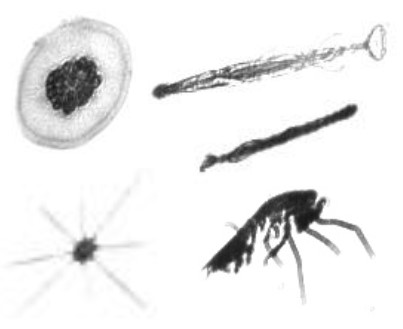 Born out of a Kaggle competition, classifying ocean plankton is an important step to be able to quantify the health of oceans. This is essentially a large multi-class problem, with close to 120 classes of plankton to be classified, with highly skewed low-resolution image data. We apply a library of image processing techniques which can characterise the shape of one plankton from another: shape descriptors (circularity constant, Hu moments), orientation descriptors (Gabor filters, HoG vectors), feature descriptors (SIFT), and information descriptors (Fourier analysis). We follow this by a library of machine learning techniques, from support vector machines, to random forests, to neural networks. Taking inspiration from the biological domain, we created hierarchically stacked classifiers mimicking the phylogeny tree structure of plankton classes. This increased accuracies on test data, from as low as 14% on a standard SVM, to as high as 82% on stacked SVMs.
Born out of a Kaggle competition, classifying ocean plankton is an important step to be able to quantify the health of oceans. This is essentially a large multi-class problem, with close to 120 classes of plankton to be classified, with highly skewed low-resolution image data. We apply a library of image processing techniques which can characterise the shape of one plankton from another: shape descriptors (circularity constant, Hu moments), orientation descriptors (Gabor filters, HoG vectors), feature descriptors (SIFT), and information descriptors (Fourier analysis). We follow this by a library of machine learning techniques, from support vector machines, to random forests, to neural networks. Taking inspiration from the biological domain, we created hierarchically stacked classifiers mimicking the phylogeny tree structure of plankton classes. This increased accuracies on test data, from as low as 14% on a standard SVM, to as high as 82% on stacked SVMs.
| slides 1 | slides 2 | code | ↑ |